Prof.Dr. Haydar BAĞIŞ
ADYÜ Tıp Fak. Dekan V.
Tıbbi Genetik ABD Başkanı.
06.08.2025
Mitokondriyal Nörogastrointestinal Ensefalomiyopati (MNGIE) Sendromu
Mitokondriyal nörogastrointestinal ensefalomiyopati (MNGIE), gastrointestinal sistem kaslarının ilerleyici dejenerasyonuyla gastrointestinal dismotiliteye, göz kapaklarının düşmesine (ptozis) ve göz hareketlerinin kısıtlanmasına (oftalmoparezi), periferik sinirlerin dejenerasyonuyla distal kollarda ve bacaklarda duyusal değişikliklere ve güçsüzlüğe ve genel zayıflamaya (kaşeksi) neden olan nadir bir multisistem bozukluğudur. MNGIE ile ilişkili spesifik semptomlar vakadan vakaya değişir ve kusma, bulantı, ishal, karın ağrısı ve ellerde ve ayaklarda uyuşma veya iğne batması hissini içerebilir. Bazı vakalarda ek bulgular ortaya çıkabilir. MNGIE, timidin fosforilazını (TP) kodlayan TYMP genindeki değişikliklerden (mutasyonlardan) kaynaklanır ve otozomal resesif bir özellik olarak kalıtılır.
MNGIE hastalarında ayrıca mitokondrilerin genetik materyalinde (DNA) değişiklikler (örneğin tükenmeler, silinmeler veya nokta mutasyonları) görülür. Vücudun hemen hemen her hücresinde yüzlercesi bulunan mitokondriler, hücresel enerjinin çoğunu, şekerlerden ve yağlardan elde edilen elektronları hücrenin enerji birimi olan ATP'ye dönüştüren solunum zinciri enzimleri (kompleks IV) aracılığıyla üretir. Solunum zincirinin temel bileşenlerinin genetik planları mitokondriyal DNA'dır (mtDNA). Genellikle solunum zincirindeki kusurlardan kaynaklanan mitokondriyal işlev bozukluğuna bağlı bozukluklara mitokondriyal hastalık denir. Enerji birçok doku işlevi için gerekli olduğundan, mitokondriyal hastalıklar genellikle vücudun birden fazla organını etkiler.
Belirtiler ve Semptomlar
MNGIE'nin semptomları ve şiddeti vakadan vakaya değişir. Semptomların başlangıcı genellikle 20 yaşından öncedir, ancak 5-60 yaşları arasında da değişebilir. MNGIE, çeşitli gastrointestinal ve nörolojik bulgularla karakterizedir.
MNGIE'nin en belirgin belirtisi, gastrointestinal sistem kaslarının ilerleyici işlev bozukluğudur (gastrointestinal dismotilite). Boğazın arkasından (orofarenks) kalın bağırsağa kadar gastrointestinal sistemin herhangi bir bölümü etkilenebilir. MNGIE ile ilişkili en yaygın gastrointestinal dismotilite türü, bağırsak duvarlarındaki kasların normal şekilde kasılmadığı ve yiyecekleri sindirim sisteminden itmek için gereken dalga benzeri (peristaltik) hareketleri üretemediği ince bağırsak hipomotilitesidir. Fiziksel bir tıkanıklık olmamasına rağmen yiyeceklerin bağırsaklardan itilememesi, intestinal psödoobstrüksiyon olarak bilinir. Mide duvarı kasları etkilendiğinde ise duruma gastroparezi denir.
MNGIE'li bireylerde çeşitli gastrointestinal semptomlar gelişebilir. Spesifik semptomlar vakadan vakaya değişmekle birlikte kusma, mide bulantısı, ishal, karın ağrısı, erken tokluk hissi (erken doyma), mide guruldaması (borborigmi) ve yutma güçlüğü (disfaji) gibi semptomlar içerebilir. Etkilenen bazı bireylerde ayrıca, ince bağırsağın dış kas duvarından iç bağırsak tabakasında küçük kese benzeri çıkıntılar (divertiküller) gelişebilir.
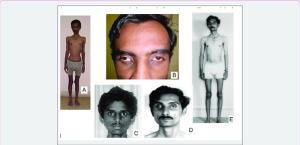
MNGIE ile ilişkili gastrointestinal semptomlar, bağırsaklarda bakteriyel aşırı çoğalma ve daha az sıklıkla bağırsakların sindirim sırasında besinleri emememesi (malabsorpsiyon) dahil olmak üzere çeşitli komplikasyonlara yol açabilir. Sonuç olarak, etkilenen bireylerde kilo kaybı ve doku ve kas kütlesi kaybı (kaşeksi) görülebilir. MNGIE'li bireylerin çoğu aşırı zayıftır ve bazılarında boy kısalığı görülebilir.
MNGIE ile ilişkili en yaygın nörolojik semptomlar, göz kapağı kaslarının zayıflığına bağlı üst göz kapağının düşmesi (ptozis), göz çevresindeki ek kasların zayıflığı nedeniyle gözlerin hareketlerinin giderek kısıtlanması (oftalmopleji), işitme kaybı ve periferik nöropatidir. Periferik nöropati, periferik sinir sisteminin (yani merkezi sinir sistemi dışındaki sinirlerin) hasar görmesi veya işlev bozukluğu yaşamasıdır. Periferik nöropatinin semptomları büyük ölçüde değişiklik gösterir, ancak distal kol veya bacak kaslarında güçsüzlük veya karıncalanma (parestezi), yanma veya uyuşma gibi anormal hisler içerebilir. Bacaklar, kollardan daha sık ve daha erken etkilenir.
MNGIE'li bireylerde sıklıkla beyindeki sinir liflerini kaplayan miyelin kılıfında hasar (lökoensefalopati) görülür. Bu klinik bulgu genellikle semptomlarla ilişkili değildir (asemptomatik).
Nedenler
MNGIE, otozomal resesif olarak kalıtılır. Genetik hastalıklar, babadan ve anneden alınan kromozomlardaki belirli bir karaktere ait genlerin kombinasyonuyla belirlenir.
Resesif genetik bozukluklar, bir bireyin her iki ebeveyninden de aynı özellik için aynı anormal geni miras almasıyla ortaya çıkar. Bir birey bir normal gen ve bir hastalık geni alırsa, kişi hastalık taşıyıcısı olur, ancak genellikle semptom göstermez. Taşıyıcı ebeveynlerden ikisinin de kusurlu geni aktarması ve dolayısıyla hastalıklı bir çocuğa sahip olma riski her hamilelikte %25'tir. Ebeveynleri gibi taşıyıcı bir çocuğa sahip olma riski her hamilelikte %50'dir. Bir çocuğun her iki ebeveyninden de normal genler alıp o özellik için genetik olarak normal olma olasılığı %25'tir. Risk hem erkekler hem de kadınlar için aynıdır.
MNGIE, timidin fosforilazını (TP) kodlayan TYMP genindeki mutasyonlardan kaynaklanır; gen, 22. kromozomun (22q13.32-qter) uzun kolunun (q) ucuna (telomer) yakın bir yerde bulunur. İnsan hücrelerinin çekirdeğinde bulunan kromozomlar, her bir birey için genetik bilgiyi taşır. İnsan vücut hücreleri normalde 46 kromozoma sahiptir. İnsan kromozom çiftleri 1'den 22'ye kadar numaralandırılır ve cinsiyet kromozomları X ve Y olarak adlandırılır. Erkeklerde bir X ve bir Y kromozomu, kadınlarda ise iki X kromozomu bulunur. Her kromozomun "p" olarak adlandırılan kısa bir kolu ve "q" olarak adlandırılan uzun bir kolu vardır. Kromozomlar ayrıca numaralandırılmış birçok banda ayrılır. Örneğin, "kromozom 22q13.32-qter" ifadesi, kromozom 22'nin uzun kolunun uç kısmına (ter) kadar olan bantları ifade eder. Numaralandırılmış bantlar, her kromozomda bulunan binlerce genin yerini belirtir.
TYMP geni, vücuttaki belirli kimyasal bileşiklerin (nükleozidler) parçalanması ve dönüştürülmesi için gerekli bir enzim olan timidin fosforilaz üretimi için talimatlar içerir (kodlar). TP genindeki mutasyonlar, vücudun hücre ve dokularında timidin fosforilaz aktivitesinin çok düşük olmasına neden olur. TP enziminin eksikliği, vücutta anormal derecede yüksek nükleozid, deoksiuridin ve timidin seviyelerine neden olur. Araştırmacılar, yüksek timidin seviyelerinin mitokondriyal DNA replikasyonunu, onarımını veya her ikisini birden bozduğuna veya hasar verdiğine inanmaktadır. MNGIE'li bireylerde mitokondriyal DNA kusurları vardır (örneğin, tükenmeler, silinmeler, çoğalmalar). Mitokondriyal DNA yaşam boyunca sürekli olarak onarılır ve çoğalır, bu nedenle etkilenen bireylerin yaşlandıkça ek mitokondriyal DNA anormallikleri biriktirdiğine inanılmaktadır.
Etkilenen nüfuslar
MNGIE, erkekleri ve kadınları eşit oranda etkileyen son derece nadir bir hastalıktır. Yaygınlığı bilinmemektedir ve 2011 itibarıyla tıp literatüründe 200'den az vaka bildirilmiştir. Araştırmacılar, hastalığın genellikle fark edilmediğine veya yanlış teşhis edildiğine, bu nedenle genel popülasyondaki gerçek sıklığını belirlemenin zor olduğuna inanmaktadır.
MNGIE, tıp literatüründe ilk olarak 1976 yılında anormal kas ve karaciğer mitokondrileriyle seyreden konjenital oküloskeletal miyopati olarak tanımlanmıştır. O zamandan beri, sensörimotor polinöropati, oftalmopleji ve psödoobstrüksiyonlu mitokondriyal ensefalomiyopati (MEPOP); miyonörogastrointestinal ensefalopati sendromu; okülogastrointestinal kas distrofisi; ve polinöropati, oftalmopleji, lökoensefalopati ve intestinal psödoobstrüksiyon (POLIP) dahil olmak üzere çeşitli isimler altında bildirilmiştir.
Benzer Belirtilere Sahip Bozukluklar
Aşağıdaki bozuklukların belirtileri MNGIE'ye benzer olabilir. Karşılaştırmalar ayırıcı tanı için faydalı olabilir.
Mitokondriyal bozukluklar, hücrenin enerji salgılayan kısımlarını (mitokondri) etkileyen mutasyonlarla karakterize bir grup ilişkili bozukluktur. Mitokondriyal bozukluklar genellikle etkilenen hücrelerin enerji üretmek için besinleri oksijenle birleştirme yeteneğini engeller. Çoğu mitokondriyal bozuklukta, vücut hücrelerinde anormal derecede yüksek sayıda kusurlu mitokondri bulunur. Mitokondriyal hastalıklar genellikle vücuttaki birden fazla organ sistemini etkiler. Mitokondriyal bozukluklarla ilişkili yaygın semptomlar arasında kas güçsüzlüğü, felç benzeri ataklar ve nöbetler bulunur. Egzersiz intoleransı da yaygın bir semptomdur. Bazı formları kalp kası hastalığı (kardiyomiyopati) ile ilişkilidir. Mitokondriyal bozukluklar arasında Kearns-Sayre sendromu, MELAS sendromu, MERRF sendromu, NARP ve Leber kalıtsal optik nöropatisi bulunur. (Bu bozukluk hakkında daha fazla bilgi için, Nadir Hastalıklar Veritabanı'nda arama teriminiz olarak belirli bozukluk adını seçin.)
Lökodistrofiler, beyni, omuriliği ve sıklıkla periferik sinirleri etkileyen çok nadir, ilerleyici, metabolik ve genetik bir hastalık grubudur. Her bir lökodistrofi türü, beynin beyaz cevherini (miyelin kılıfı) oluşturan en az 10 farklı kimyasaldan birinin anormal gelişimine yol açan belirli bir gen anormalliğinden kaynaklanır. Miyelin kılıfı, sinirin koruyucu kılıfıdır ve sinirler onsuz normal şekilde çalışamaz. Her bir lökodistrofi türü, miyelin kılıfının farklı bir bölümünü etkileyerek bir dizi nörolojik soruna yol açar. Lökodistrofi, hareket, görme, işitme, denge, yeme yeteneği, hafıza, davranış ve düşünce ile ilgili sorunlara neden olabilir. Lökodistrofiler ilerleyici hastalıklardır, yani hastalığın belirtileri zamanla kötüleşme eğilimindedir. (Bu hastalık hakkında daha fazla bilgi için Nadir Hastalıklar Veritabanı'nda arama teriminiz olarak "lökodistrofi"yi seçin.)
Crohn hastalığı da dahil olmak üzere birçok hastalık, MNGIE'de bulunanlara benzer gastrointestinal semptomlara sahiptir. Crohn hastalığı, bağırsak duvarında veya gastrointestinal sistemin herhangi bir bölümünde şiddetli, kronik iltihaplanma ile karakterize bir inflamatuar bağırsak hastalığıdır. İnce bağırsağın alt kısmı (ileum) ve rektum bu hastalıktan en sık etkilenen bölgelerdir. Semptomlar arasında sulu ishal, karın ağrısı, ateş ve kilo kaybı yer alabilir. Crohn hastalığının semptomlarının yönetimi zor olabilir ve doğru teşhis genellikle gecikir. Crohn hastalığının kesin nedeni bilinmemektedir. (Bu hastalık hakkında daha fazla bilgi için Nadir Hastalıklar Veritabanı'nda arama terimi olarak "Crohn"u seçin.)
Tanı
MNGIE tanısı, ayrıntılı hasta öyküsü, kapsamlı bir klinik değerlendirme, karakteristik bulguların belirlenmesi ve kan testleri veya manyetik rezonans görüntüleme (MRG) gibi çeşitli özel testlere dayanarak şüphelenilir. Kan testleri laktik asit yüksekliği gösterebilir. MR, belirli organların ve vücut dokularının kesitsel görüntülerini oluşturmak için manyetik alan ve radyo dalgaları kullanır. MNGIE'li bireylerde, asemptomatik lökoensefalopatiyi göstermek için kullanılır.
MNGIE tanısı, beyaz kan hücreleri (lökositler) ve trombositleri içeren buffy coat'ta düşük TP enzim aktivitesi gösterilerek veya plazmada timidin ve deoksiüridin nükleozitlerinin yüksek seviyelerinin tespit edilmesiyle biyokimyasal olarak doğrulanabilir. Alternatif olarak, deoksiribonükleik asit (DNA) incelemesinin MNGIE ile ilişkili spesifik genetik mutasyonları ortaya çıkardığı moleküler genetik testlerle de tanı doğrulanabilir.
Standart Terapiler
Tedavi
MNGIE tedavisi, her bireyde görülen spesifik semptomlara yöneliktir. Tedavi, uzmanlardan oluşan bir ekibin koordineli çalışmasını gerektirebilir. Çocuk doktorları, iç hastalıkları uzmanları, gastroenterologlar, nörologlar, cerrahlar, kardiyologlar, diş hekimleri, konuşma patologları, işitme sorunlarını değerlendiren ve tedavi eden uzmanlar (odyologlar), göz uzmanları ve diğerleri, etkilenen hastanın tedavisini sistematik ve kapsamlı bir şekilde planlamak zorunda kalabilir.
Spesifik tedaviler arasında bulantı ve kusma için ilaç tedavileri ve ağrı gibi sinir disfonksiyonuna (nöropati) bağlı semptomlar yer alabilir. Gastrointestinal sistemin belirli organlarını (karın içi organları) etkileyen ağrı, çölyak pleksus nörolizi olarak bilinen bir işlemle tedavi edilebilir. Bu işlem sırasında sinir uyarıları geçici olarak kesilerek ağrı azaltılır.
Etkilenen bireylerin aspirasyonu önlemek için yutma güçlüğü açısından değerlendirilmesi gerekebilir.
Besin takviyesi gerekebilir ve parenteral beslenme veya gastrostomi tüpü yoluyla sağlanabilir. Parenteral beslenme, gastrointestinal sistem veya akciğerleri içermeyen herhangi bir yoldan beslenmedir; yani bir tüp aracılığıyla doğrudan damarlara (intravenöz) beslenmedir. Gastrostomi, midede cerrahi bir açıklık oluşturulması ve bu açıklıktan doğrudan beslenme desteği sağlamak için bir tüpün yerleştirilmesi anlamına gelir.
MNGIE'li bireyler, mitokondriyal işlevi etkileyen veya engelleyen ilaçlardan kaçınmalıdır. Bu ilaçlar arasında valproat, fenitoin, kloramfenikol, linezolid, aminoglikozitler ve tetrasiklin bulunur.
Etkilenen bazı bireyler mesleki terapi ve fizik tedaviden fayda görebilir. Genetik danışmanlık, etkilenen bireyler ve aileleri için faydalı olabilir. Diğer tedaviler semptomatik ve destekleyicidir.
Klinik Araştırmalar ve Çalışmalar
Araştırmacılar, MNGIE'li bireylerin hücre ve dokularındaki nükleozit düzeylerini düşürmeyi amaçlayan çeşitli tedavi seçenekleri üzerinde çalışmaktadır. Bu tedaviler arasında diyaliz ve allojenik kök hücre (kemik iliği) nakli yer almaktadır. Bu tedaviler henüz erken araştırma aşamasındadır ve önemli dezavantajlar ve riskler taşımaktadır. Araştırma aşamasında olan bir diğer tedavi seçeneği ise böbrekler tarafından timidin emilimini azaltarak idrar yoluyla daha fazla timidin atılmasını sağlamaktır. Bu potansiyel tedavilerin MNGIE'li bireyler için uzun vadeli güvenliğini ve etkinliğini belirlemek için daha fazla araştırmaya ihtiyaç vardır.
Mevcut klinik araştırmalara ilişkin bilgiler internette www.clinicaltrials.gov adresinde yayınlanmaktadır.
Bu Hastalığın Diğer İsimleri
MEPOP
Mitokondriyal nörogastrointestinal ensefalopati sendromu
MNGIE hastalığı
MNGIE sendromu
Miyonörogastrointestinal ensefalopati sendromu
Okülogastrointestinal kas distrofisi
Timidin fosforilaz eksikliği
KAYNAKLARh
ttps://rarediseases.org/rare-diseases/mitochondrial-neurogastrointestinal-encephalopathy/
https://medlineplus.gov/genetics/condition/mitochondrial-neurogastrointestinal-encephalopathy-disease/#references
Halter J, Schüpbach WM, Casali C, et al. Allogeneic hematopoietic SCT as treatment option for patients with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): a consensus conference proposal for a standardized approach. Bone Marrow Transplant. 2011;46:330-337.
Garone C, Tadesse S, Hirano M. Clinical and genetic spectrum of mitochondrial neurogastrointestinal encephalomyopathy. Brain. 2011;134:326-332.
Lara MC, Weiss B, Illa I, et al. Infusion of platelets transiently reduces nucleoside overload in MNGIE. Neurology. 2006;67:1461-3.
Hirano M, Marti R, Casali C, et al. Allogenic stem cell transplantation corrects biochemical derangements in MNGIE. Neurology. 2006;67:1458-60.
Marti R, Verschuuren JJ, Buchman A, et al. Late-onset MNGIE due to partial loss of thymidine phosphorylase deficiency. Ann Neurol. 2005;58:649-52.
Szigeti K, Wong LJC, Perng CL, et al. MNGIE with lack of skeletal muscle involvement and a novel TP splice site mutation. J Med Genet. 2004;41:125-9.
Spinazzola A, Marti R, Nishino I, et al. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2002;6:4128-33.
Teitelbaum JE, Berde CB, Nurko S, et al. Diagnosis and management of MNGIE syndrome in children: case report and review of the literature. J Pediatr Gastroenterol Nutr. 2002;35:377-83.
Spinazzola A, Marti R, Nishino I, et al. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem. 2001;277:4128-33.
Nishino I, Spinazzola A, Papadimitriou A, et al. Mitochondrial neurogastrointestinal encephalomyopathy: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol. 2000;47:792-800.
Hirano M, Garcia-de-Yebenes J, Jones AC, et al. Mitochondrial neurogastrointestinal encephalomyopathy syndrome maps to chromosome 22q13.32-qter. Am J Hum. 1998;63:526-33.
Bardosi A, Creutzfeldt W, DiMauro S, et al. Myo-,neuro-, gastrointestinal encephalopathy (MNGIE syndrome) due to partial deficiency of cytochrome-c-oxidase. Acta Neuropathol. 1987.74:248-58.
Ionasescu V. Oculogastrointestinal muscular dystrophy. Am J Med Genet. 1983;15:103-12.
Anuras S, Mitros FA, Nowak TV. A familial visceral myopathy with external ophthalmoplegia and autosomal recessive transmission. Gastroenterology. 1983;84:346-53.