Prof.Dr. Haydar BAĞIŞ
ADYÜ Tıp Fak. Dekan V.
Tıbbi Genetik ABD Başkanı.
14.04.2025
Prader-Willi Sendromu
Prader-Willi sendromu, vücudun birçok bölümünü etkileyen karmaşık bir genetik durumdur. Bebeklikte, bu durum zayıf kas tonusu (hipotoni), beslenme zorlukları, zayıf büyüme ve gecikmiş gelişim ile karakterizedir. Çocukluktan başlayarak, etkilenen bireylerde aşırı açlık gelişir ve bu da kronik aşırı yeme (hiperfaji) ve obeziteye yol açar. Prader-Willi sendromu olan bazı kişilerde, özellikle obezitesi olanlarda, tip 2 diyabet (diyabetin en yaygın biçimi) de gelişir.
Prader-Willi sendromu olan kişilerde genellikle hafif ila orta düzeyde zihinsel bozukluk ve öğrenme güçlükleri görülür. Öfke patlamaları, inatçılık ve cildi yolma gibi zorlayıcı davranışlar gibi davranışsal sorunlar yaygındır. Uyku bozuklukları da görülebilir. Bu durumun ek özellikleri arasında dar alın , badem şeklinde gözler ve üçgen ağız gibi belirgin yüz özellikleri; kısa boy; ve küçük eller ve ayaklar bulunur . Prader-Willi sendromu olan bazı kişilerde alışılmadık derecede açık ten ve açık renkli saçlar bulunur . Hem etkilenen erkeklerde hem de etkilenen kadınlarda az gelişmiş cinsel organlar vardır. Ergenlik gecikir veya eksiktir ve etkilenen bireylerin çoğu çocuk sahibi olamaz (kısırlık).
Nedenler
Prader-Willi sendromu , kromozom 15'in belirli bir bölgesindeki genlerin işlevini kaybetmesinden kaynaklanır . İnsanlar normalde bu kromozomun bir kopyasını her ebeveynden miras alırlar. Bazı genler yalnızca kişinin babasından miras alınan kopyada (paternal kopya) açılır (aktifleşir). Bu ebeveyne özgü gen aktivitesi, genomik baskılama adı verilen bir süreçten kaynaklanır.
Prader-Willi sendromunun çoğu vakası (yaklaşık %70'i), her hücrede paternal kromozom 15'in bir segmentinin silinmesiyle meydana gelir. Bu kromozomal değişikliğe sahip kişilerde, paternal kopyadaki genler silindiği ve maternal kopyadaki genler kapatıldığı (etkisiz hale getirildiği) için bu bölgedeki belirli kritik genler eksiktir. Vakaların bir diğer %25'inde, Prader-Willi sendromu olan bir kişinin her bir ebeveynden birer kopya yerine annesinden miras aldığı iki kromozom 15 kopyası (maternal kopyalar) vardır. Bu duruma maternal uniparental dizomi (UPD) denir. Nadiren, Prader-Willi sendromuna translokasyon adı verilen bir kromozomal yeniden düzenleme veya paternal kromozom 15'teki genleri anormal şekilde kapatan (etkisiz hale getiren) bir genetik değişiklik veya başka bir değişiklik de neden olabilir .
Prader-Willi sendromunun karakteristik özelliklerinin kromozom 15'teki birkaç genin işlevini kaybetmesinden kaynaklandığı muhtemeldir. Bunlar arasında küçük nükleolar RNA'lar (snoRNA'lar) adı verilen moleküllerin yapımına yönelik talimatlar sağlayan genler de vardır. Bu moleküllerin, diğer RNA molekülü tiplerini düzenlemeye yardımcı olmak da dahil olmak üzere çeşitli işlevleri vardır. (RNA molekülleri protein üretmede ve diğer hücre aktivitelerinde önemli roller oynar.) Çalışmalar, SNORD116 kümesi olarak bilinen belirli bir snoRNA gen grubunun kaybının, Prader-Willi sendromunun belirti ve semptomlarına neden olmada önemli bir rol oynayabileceğini öne sürmektedir. Ancak, eksik bir SNORD116 kümesinin zihinsel engelliliğe, davranış sorunlarına ve bozukluğun fiziksel özelliklerine nasıl katkıda bulunabileceği bilinmemektedir.
Prader-Willi sendromu olan bazı kişilerde, OCA2 adı verilen bir genin kaybı alışılmadık derecede açık ten ve açık renkli saçlarla ilişkilidir . OCA2 geni, bu bozukluğa sahip kişilerde sıklıkla silinen kromozom 15 segmentinde yer alır. Ancak, OCA2 geninin kaybı Prader-Willi sendromunun diğer belirti ve semptomlarına neden olmaz. Bu genden üretilen protein, cildin, saçın ve gözlerin rengini (pigmentasyonunu) belirlemeye yardımcı olur.
KALITIMI
Prader-Willi sendromunun çoğu vakası kalıtsal değildir, özellikle paternal kromozom 15'teki bir delesyon veya maternal uniparental disomi nedeniyle oluşanlar . Bu genetik değişiklikler üreme hücrelerinin (yumurta ve sperm) oluşumu sırasında veya erken embriyonik gelişimde rastgele olaylar olarak meydana gelir. Etkilenen kişilerin ailelerinde genellikle bu bozuklukla ilgili bir geçmiş yoktur.
Nadiren, Prader-Willi sendromundan sorumlu genetik bir değişiklik kalıtsal olabilir. Örneğin, baba kromozomu 15'teki genleri anormal şekilde kapatan bir genetik değişikliğin bir nesilden diğerine geçmesi mümkündür.
Ayırıcı tanı
Tanı sürecinde PWS'ye güçlü şekilde benzeyebilen birkaç bozukluk dikkate alınmalıdır. İnfantil hipotoninin, santral ve periferik, sendromik ve izole olmak üzere çok uzun bir neden listesi vardır. Tanı FISH ile bir bebekte konulmuşsa Angelman sendromu (AS) düşünülmelidir, çünkü AS ayrıca hipotoni ve beslenme zorluklarıyla da ortaya çıkar. AS ayrıca sıklıkla 15q11.2–q13 delesyonundan (70%) kaynaklanır, ancak maternal 15'te. Bu nedenle, delesyonu olan 2 yaşından küçük tüm bebeklerde AS ve PWS'yi ayırt etmek için DNA metilasyon testi yapılmalıdır.
Fragile X sendromlu bireylerin bir alt kümesi hiperfajik ve obez olabilir. UPD 14, Cohen sendromu, Bardet-Biedl sendromu, Alstrom sendromu, 3p25.3–p26.2 ve Xq27.2-ter duplikasyonları ve 1p36, 6q16.2 ve 10q26 delesyonları dahil olmak üzere bir dizi başka durum da obezite ve gelişimsel engelliliği ilişkilendirir.
Sendromun Diğer İsimleri
Prader-Labhart-Willi sendromu
PWS
Willi-Prader sendromu
Uyku bozuklukları
PWS'li bireylerde, merkezi ve obstrüktif uyku apnesi, anormal uyarılma, REM uykusunda anormal sirkadiyen ritim, azalmış REM latensi ve hiperkapniye anormal yanıt ve ayrıca aşırı gündüz uykululuğu gibi uykuda solunum bozukluğu görülür. Obezite uyku bozukluğunu kötüleştirebilir.
Ölüm oranı
PWS'deki ölümlerle ilgili birkaç rapor ve büyük kohortlardaki anketler, obeziteyle ilişkili kardiyovasküler ve solunum bozukluklarının hem çocuklarda hem de yetişkinlerde en sık görülen ölüm nedenleri olduğunu bulmuştur. Bir nüfus çalışmasına dayanarak, ölüm oranının yılda %3 olduğu tahmin edilmektedir. Başka bir büyük çalışma, PWS'de diğer gelişimsel engelli bireylere kıyasla 6 kat daha fazla ölüm riski olduğunu ileri sürmüştür. Aşırı yemekten boğulma ve özellikle zayıf ancak önceden obez olan bireylerde aşırı yeme sonrası mide nekrozu ve yırtılması gibi yemeyle ilişkili ölümler konusunda özel endişeler dile getirilmiştir. Son zamanlarda, test edilen PWS'li bireylerin %60'ında merkezi adrenal yetmezliğin (CAI) mevcut olduğu bulunmuştur ve yazarlar stres zamanlarında CAI'nin PWS'deki ani ve açıklanamayan ölümlerin çoğunu açıklayabileceğini öne sürmektedir.
Prader-Willi sendromunun tedavisi var mı?
Şu anda PWS için bir tedavi yoktur ve bugüne kadar yapılan araştırmaların çoğu belirli semptomları tedavi etmeye yöneliktir. Bozukluktan etkilenen birçok kişi için sendromun en zor yönlerinden bazılarının, örneğin doymak bilmez iştah ve obezitenin ortadan kaldırılması, yaşam kalitesinde ve bağımsız yaşama becerisinde önemli bir iyileşme sağlayacaktır. Prader-Willi Araştırma Vakfı, PWS için nihai bir tedavi hedefiyle yeni tedavilere yol açacak araştırmaları ilerletmektedir. PWS'nin belirli yönlerini tedavi etmek için ilaçları değerlendirmek üzere bir dizi klinik çalışma devam etmektedir ve FPWR , PWS için genetik terapiyi araştıran çalışmaları desteklemektedir .
PWS MEKANİZMA
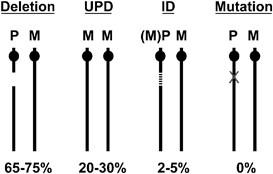
PWS'nin moleküler sınıfları ve sıklıkları. 15q11.2–q13 delesyonu, maternal uniparental dizomi (UPD), baskı defekti (ID) ve tek gen mutasyonu. Her kromozomun kökeninin ebeveyni M (maternal) veya P (paternal) ile gösterilir. ID durumunda iki kromozom 15'in biparental katkısı olduğunu, ancak 15q11.2–q13'teki paternal (P) katkısının maternal (M) epigenetik işaretlere (örneğin, DNA metilasyonu) sahip olduğunu ve buna göre davrandığını unutmayın.
KAYNAKLAR
https://medlineplus.gov/genetics/condition/prader-willi-syndrome/#references
Picture: https://www.nature.com/articles/ejhg2008165:
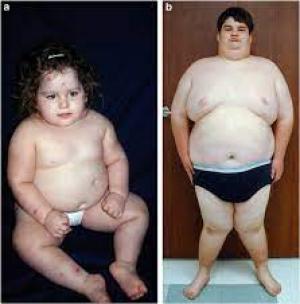
2 ½ yaşında bir kız ve 21 yaşında bir erkekte PWS'nin merkezi obezitesi . Ayrıca kızda tipik yüz görünümüne, aktif deri yolma lezyonlarına ve gastrostomi beslenme tüpü yarasına, erkekte ise psödojinekomasti ve genus valgus'a dikkat edin.