Prof.Dr. Haydar BAĞIŞ
29 Ekim 2022
Nörofibromatoz tip 1 (NF1), cilt rengindeki değişiklikler (pigmentasyon) ve cilt, beyin ve vücudun diğer kısımlarındaki sinirler boyunca tümörlerin büyümesi ile karakterize bir durumdur. Bu durumun belirti ve semptomları, etkilenen insanlar arasında büyük farklılıklar gösterir.
NF 1, dünya çapında 3.000 ila 4.000 kişiden 1'inde görülür.
NF1 kromozom 17q11.2 bölgesinde bulunan neurofibromin genindeki (NF1) heterozigot mutasyonlar sorumludur. NF1 geni 58 ekzondan ve 12425 baz çiftinden oluşan oldukça büyük bir gen olup, bugüne kadar NF 1 olgularında farklı mutasyonlar belirlenmiştir.
NF1 genindeki mutasyonlar tip 1 nörofibromatozise neden olur. NF1 geni, nörofibromin adı verilen bir proteinin yapılması için kodlama yapar. Bu protein, sinir hücreleri ve sinirleri çevreleyen özel hücreler ( oligodendrositler ve Schwann hücreleri) dahil olmak üzere birçok hücrede üretilir. Bu özel hücreler, belirli sinir hücrelerini izole eden ve koruyan yağlı kaplamalar olan miyelin kılıflarını oluşturur.
Nörofibromin, bir tümör baskılayıcı görevi görür, yani hücrelerin çok hızlı veya kontrolsüz bir şekilde büyümesini ve bölünmesini engeller.
NF1 tam penetrans ve değişken ekspresivite gösteren otozomal dominant kalıtımlı bir hastalık olup, antisipasyon bildirilmemektedir. Özellikle aynı ailedeki bireylerde bile çok değişik klinik bulguların olabilmesi nedeniyle, hastalığın izlemi ve genetik danışmanlıkta zorluklar oluşabilmektedir.
NF1 genindeki mutasyonlar, hücre büyümesini ve bölünmesini düzenleyemeyen, nörofibrominin işlevsel olmayan bir versiyonunun üretilmesine yol açar. Sonuç olarak, nörofibromlar gibi tümörler vücuttaki sinirler boyunca oluşabilir.
NF1'deki gen mutasyonların nasıl olduğu belirsizdir ve kafe-au-lait (ciltte sütlü kahve) lekeleri ve öğrenme güçlükleri gibi nörofibromatoz tip 1'in diğer özelliklerine yol açar.
Nörofibromin, bir tümör baskılayıcı protein görevi görür. Tümör baskılayıcılar normalde hücrelerin çok hızlı veya kontrolsüz bir şekilde büyümesini ve bölünmesini engeller. Bu proteinin, hücre büyümesini ve bölünmesini uyaran başka bir proteini (ras adı verilen) kapatarak hücre aşırı büyümesini önlediği görülüyor.
Ailesel Geçiş
Nörofibromatoz tip 1'in otozomal dominant kalıtım modeline sahip olduğu kabul edilir. Bu durumdaki insanlar, her hücrede NF1 geninin mutasyona uğramış bir kopyasıyla doğarlar.
Vakaların yaklaşık yarısında, değiştirilmiş gen, etkilenen bir ebeveynden miras alınır. Kalan vakalar, NF1 genindeki yeni mutasyonlardan (de Novo) kaynaklanır ve ailelerinde hastalık öyküsü olmayan kişilerde görülür.
Her hücrede bir genin değiştirilmiş bir kopyasının bozukluğa neden olmak için yeterli olduğu diğer çoğu otozomal dominant durumun aksine, nörofibromatoz tip 1'de tümör oluşumunu tetiklemek için NF1 geninin iki kopyasının değiştirilmesi gerekir.
NF1 geni, bir kişinin yaşamı boyunca sinirleri çevreleyen özel hücrelerde meydana gelir. Bir NF1 mutasyonuyla doğan hemen hemen herkes, birçok hücrede ikinci bir mutasyon kazanır ve nörofibromatoz tip 1'in karakteristik tümörlerini geliştirir.
Erken çocukluk döneminden başlayarak, tip 1 nörofibromatozisi olan hemen hemen tüm insanlarda , cilt üzerinde çevredeki alandan daha koyu olan düz yamalar olan çok sayıda kafe-au-lait lekesi bulunur. Birey büyüdükçe bu lekelerin boyutu ve sayısı artar.
Koltuk altlarında ve kasıkta çiller tipik olarak daha sonra çocuklukta gelişir.
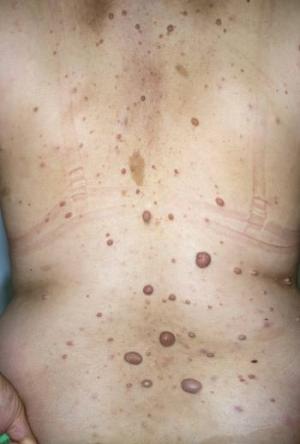
Nörofibromatoz tip 1 olan çoğu yetişkin, genellikle deri üzerinde veya hemen altında bulunan kanserli olmayan (iyi huylu) tümörler olan nörofibromlar geliştirir. Bu tümörler ayrıca omuriliğe yakın sinirlerde veya vücudun başka yerlerindeki sinirler boyunca da oluşabilir.
Nörofibromatoz tip 1 olan bazı kişilerde sinirler boyunca büyüyen kanserli tümörler gelişir. Genellikle ergenlik veya erişkinlik döneminde gelişen bu tümörlere malign periferik sinir kılıfı tümörleri denir.
Nörofibromatoz tip 1 olan kişilerde ayrıca beyin tümörleri ve kan oluşturan doku kanseri ( lösemi ) dahil olmak üzere diğer kanserleri geliştirme riski de yüksektir .
Çocukluk döneminde, Lisch nodülleri adı verilen iyi huylu büyümeler genellikle gözün renkli kısmında (iris) görülür. Lisch nodülleri görmeyi engellemez. Etkilenen bazı kişiler, gözden beyne giden sinir boyunca büyüyen tümörler de geliştirir ( optik sinir ). Optik gliomalar olarak adlandırılan bu tümörler görmede azalmaya veya tam görme kaybına neden olabilir. Bazı durumlarda, optik gliomların görme üzerinde hiçbir etkisi yoktur.
Nörofibromatoz tip 1'in ek belirti ve semptomları değişkendir, ancak bunlar arasında yüksek tansiyon ( hipertansiyon ), boy kısalığı, alışılmadık derecede büyük bir kafa ( makrosefali ) ve omurganın anormal eğriliği ( skolyoz ) gibi iskelet anormallikleri sayılabilir.
Nörofibromatozis tip 1 olan çoğu insan normal zekâya sahip olsa da, etkilenen bireylerde öğrenme güçlüğü ve dikkat eksikliği/hiperaktivite bozukluğu (DEHB) sıklıkla görülür.
Nörofibromatoz Tip 1 Belirtileri ve Belirtileri Nelerdir?
Nörofibromatozis tip 1'li yenidoğanların çoğunda semptom görülmez, ancak bazılarında kavisli alt bacak kemikleri bulunur.
İlk doğum günlerinde, NF1'li çocukların çoğunda, renkleri nedeniyle café-au-lait ("sütlü kahve") lekeleri olarak adlandırılan birkaç cilt lekesi bulunur. Ciltte sütlü kahve lekeleri noktaları şunlardır:
- Çevreleyen deriden daha koyu
- 5 milimetreden fazla
NF1'li çocukların yürümesi, konuşması ve diğer dönüm noktalarına ulaşması çoğu çocuktan daha yavaş olabilir. Ayrıca şunlara sahip olabilirler:
- Derinin içinde ve altında şişlikler ( nörofibromlar denir )
- Vücut için büyük görünen bir kafa
- Baş ağrısı
- Öğrenme zorluğu
- Dikkat sorunları ve hiperaktivite
- Aynı yaştaki çoğu çocuktan daha kısa boy
- Omurgada yan yana eğrilikler ( skolyoz )
- Önkolların veya alt bacak kemiklerinin kıvrılması, incelmesi veya zayıflığı
- Koltuk altlarında veya karın ve kalça (kasık) arasındaki kıvrımda çiller
Nörofibromlar görünüm sorunlarına neden olabilir ve ağrılı olabilir. Çok büyük nörofibromlar (pleksiform nörofibrom olarak adlandırılır) kansere dönüşebilir. NF1 semptomları bazı çocuklarda hafif, bazılarında ise şiddetlidir.
NF1 vakalarının yılda en az 1 kez, NF1 konusunda deneyimli bir nörolog tarafından muayene edilmesi; erken çocukluk döneminde yılda bir kez, sonrasında 2 yılda bir göz doktoru tarafından değerlendirme yapılması gerekir.